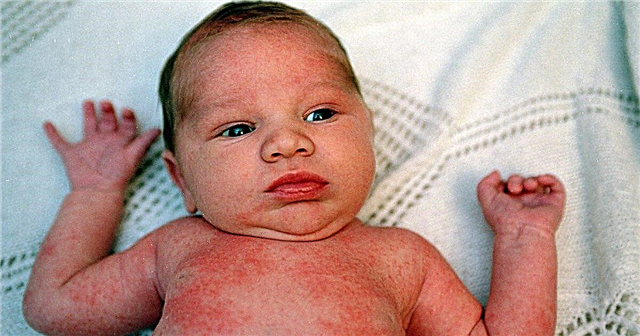
Apa itu epidermolisis bullosa?
Epidermolysis bullosa merangkumi sebilangan besar penyakit kulit yang diwarisi dan mempunyai ciri umum - lepuh dengan saiz dan bilangan yang berbeza. Mereka terbentuk pada kulit atau selaput lendir kerana kesan terkena, kadang-kadang minimum. Sebarang geseran, sentuhan, perubahan suhu dan kelembapan udara dapat memperburuk proses.
Penyakit ini dapat menampakkan diri pada usia yang berbeza. Anak menjadi sakit ketika masih dalam kandungan. Selalunya, dia sudah dilahirkan dengan tanda-tanda penyakit, atau mereka muncul sejurus selepas kelahiran. Dalam semua kes, adalah lebih tepat untuk memanggil penyakit ini sebagai kongenital epidermolysis bullosa.
Ini adalah penyakit genetik, iaitu, ia tidak menular dari orang yang sakit dalam keadaan apa pun dan dengan cara apa pun. Bayi hanya boleh mewarisinya dari ibu dan ayah pada waktu pembuahan.
Berapakah bilangan orang yang menghidap bulosa epidermolisis?
Selalunya, penyakit ini direkodkan pada kanak-kanak berumur 1 hingga 5 tahun. Data dari National Epidermolysis Bullosa Registry, yang dilakukan di Amerika Syarikat, menunjukkan bahawa 1 dari 50,000 bayi baru lahir menderita penyakit ini. Selama 16 tahun keberadaannya di Amerika Syarikat, 3300 orang telah dikenal pasti menghidap penyakit ini.
Di Eropah, epidermolisis bullosa mempengaruhi 1 dari 30,000 bayi yang baru lahir. Jepun mempunyai prevalensi penyakit yang paling rendah, ia berlaku pada 7.8 daripada 1 juta kanak-kanak yang dilahirkan.
Malangnya, tidak ada statistik rasmi mengenai penyakit ini di Rusia. Telah diketahui bahawa di negara kita, Ukraine, Belarus dan Kazakhstan, terdapat lebih dari 150 pesakit dengan epidermolisis bullosa.
Punca epidermolisis bullosa
Penyebab penyakit serius ini adalah mutasi pada gen yang bertanggungjawab untuk sintesis protein struktur pada kulit. Akibatnya, sel-selnya kehilangan hubungan yang kuat antara satu sama lain. Kesan sedikit dari luar menyumbang kepada kerosakan pada kulit.
Kulit manusia terdiri daripada beberapa lapisan. Atas - epidermis diwakili oleh sel keratinosit. Mereka terus membelah dan, ketika tumbuh, bergerak dari lapisan yang mendasari ke lapisan atas korneum, memberikan pembaharuan pada kulit dan perlindungannya. Keratinosit dihubungkan satu sama lain oleh jambatan khas - desmosomes, dari mana filamen protein - tonofibril - menonjol. Sel-sel lapisan bawah epidermis juga dihubungkan oleh protein laminin.
Epidermis diikuti oleh lapisan yang disebut dermis. Ini termasuk kolagen, serat elastik dan retikular, yang meresap oleh banyak saluran darah dan limfa, saraf, peluh dan kelenjar sebum, folikel rambut. Dermis mengandungi sel fibroblast. Merekalah yang menghasilkan semua serat di lapisan.
Dermis dan epidermis dihubungkan dengan kuat oleh lapisan - membran bawah tanah. Ia juga mengandungi protein kolagen. Mereka adalah komponen utama benang penahan. Filamen ini melekatkan membran bawah tanah dengan erat dengan epidermis penutup ke dermis.
Gangguan struktur kulit mendasari pelbagai bentuk klinikal epidermolisis bullosa, apabila, sebagai hasil mutasi, pertumbuhan dan pematangan keratinosit yang rosak terjadi, ketiadaan atau penurunan penahanan dan pengikat filamen protein. Kekurangan protein tertentu yang terlibat dalam struktur kulit juga mungkin berlaku: kolagen, keratin, laminin dan lain-lain.
Akibatnya, hubungan antara lapisan kulit dan selnya menjadi lemah, dan sedikitpun pengaruh dari luar, kerosakan berlaku dengan pembentukan gelembung.
Klasifikasi bullosa epidermolisis
Sejak pengembangan teknologi yang memungkinkan untuk menentukan struktur kulit pada tahap mikroskopik dan menentukan struktur terkecilnya, epidermolisis bullosa telah dibahagikan kepada 3 kumpulan. Kemudian, kumpulan penyakit lain dikenal pasti.
Dalam komuniti perubatan moden, klasifikasi penyakit terdiri daripada 4 kumpulan utama dan 6 subkumpulan. Dalam subkumpulan, pelbagai bentuk klinikal penyakit dikumpulkan. Mereka berbeza dalam jenis pewarisan, perubahan mikroskopik pada kulit, manifestasi klinikal, keparahan, prognosis.
Jadi, bulosa epidermolisis boleh menjadi sederhana, sempadan, distrofik. Sindrom Kindler adalah kumpulan yang berasingan. Bullosa epidermolisis sederhana merangkumi suprabasal dan basal. Bentuk bulosa epidermolisis sempadan dapat dilokalisasi dan digeneralisasikan. Bullosa epidermolisis distrofi dominan dan berulang. Sindrom Kindler tidak terbahagi kepada subkumpulan.
Pengelasannya mengambil kira lapisan kulit di mana pundi kencing terbentuk, dan juga perubahan protein yang membentuknya.
Manifestasi bulosa epidermolisis
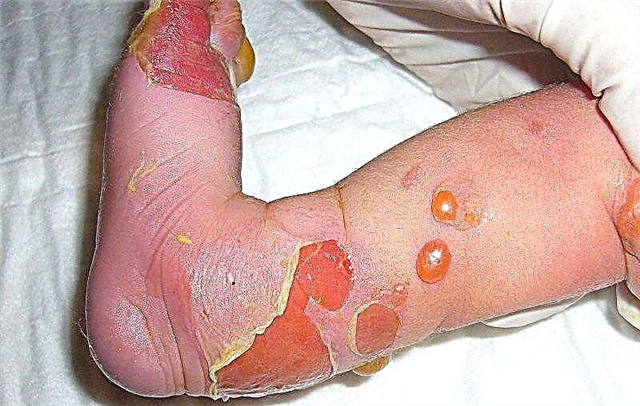
Lepuh, erosi pelbagai saiz pada kulit dan membran mukus adalah tanda utama epidermolisis bullosa. Mereka muncul kerana penurunan daya tahan kulit terhadap pelbagai pengaruh dari persekitaran luaran. Ini sering berlaku apabila suhu persekitaran berubah, kesan tekanan dan geseran. Oleh kerana kulit mempunyai struktur yang tidak biasa, gelembung muncul, kemudian hakisan. Penyembuhan mereka dengan beberapa jenis bulosa epidermolisis dapat berlaku dengan pembentukan parut kasar.
Manifestasi lain dari epidermolisis bullosa sering merangkumi perubahan warna kulit, keratosis pada telapak tangan dan tapak kaki, kontraksi sendi kecil tangan, dan peleburan jari. Yang kurang biasa adalah kebotakan, atau rambut gugur separa, peningkatan keringat, atau, sebaliknya, ketiadaannya, kerosakan gigi, kesukaran menelan, muntah, sembelit, cirit-birit.
Bullosa epidermolisis sederhana
Bayi dengan bulosa epidermolisis sederhana dilahirkan dengan lecet pada kulit, atau ia muncul pada bulan-bulan pertama kehidupan mereka. Lepuh boleh dilihat pada tangan, kaki, siku, lutut, tulang kering, kulit kepala. Di rongga mulut terdapat sedikit atau tidak sama sekali. Lepuh dan erosi tidak menyakitkan dan cepat sembuh.
Kuku tidak berubah. Sekiranya detasmen mereka berlaku, mereka mesti dipulihkan. Gigi kanak-kanak seperti itu sihat. Lepuh sembuh tanpa meninggalkan jejak. Semasa kanak-kanak itu membesar, mereka akan semakin berkurang.

Simplex epidermolisis bullosa tempatan dan tangan merujuk kepada simplex bullosa epidermolisis basal. Ia menampakkan diri dengan permulaan berjalan bebas. Penyakit ini juga boleh bermula pada remaja, apabila memakai kasut yang ketat bermula dan kaki cedera. Mereka paling kerap terkena. Kekalahan bahagian badan yang lain kurang biasa. Jumlah mereka berbeza, dari ruam kecil hingga besar, yang membatasi kehidupan biasa.
Bullosa epidermolisis umum juga merupakan salah satu bentuk klinikal bullosa epidermolisis basal. Pada bayi, bahagian belakang kepala, belakang, siku terjejas. Semasa anak tumbuh dan matang, gelembung muncul di tangan, kaki, dan tempat yang mengalami geseran.
Terdapat banyak gelembung. Mereka terletak dalam kumpulan yang membentuk pusat pelbagai garis besar pelik. Selaput lendir terjejas, kuku mudah terkeluar. Pigmentasi kulit sering berubah, hiperhidrosis dan hiperkeratosis telapak tangan dan tapak kaki muncul. Penting - selepas kerosakan kulit, tidak ada bekas luka.
Biasanya, apabila suhu persekitaran meningkat, keadaan pesakit dengan epidermolisis bullosa bertambah buruk. Walau bagaimanapun, pada bayi dengan bentuk umum epidermolisis bullosa, peningkatan suhu mempunyai kesan positif terhadap keadaan kulit.
Bullosa epidermolisis sempadan
Dengan bullosa epidermolisis sempadan, perubahan mempengaruhi membran bawah epidermis. Ia dibahagikan kepada umum dan dilokalisasikan. Ciri yang umum adalah penampilan lepuh di mana-mana bahagian badan dengan lesi yang luas. Perubahan pada enamel gigi juga menjadi ciri, ia menjadi lebih nipis, titik kemurungan muncul di permukaan gigi. Mereka terdedah kepada kerosakan gigi. Bullosa epidermolisis sempadan lebih teruk daripada sederhana.
Bullosa epidermolisis sempadan umum yang teruk juga disebut "mematikan". Ini adalah penyakit yang boleh membawa maut, hasilnya adalah cacat dan kecacatan. Bayi dilahirkan dengan gelembung, atau usia manifestasi mereka adalah tempoh neonatal. Lepuh sering dilokalisasi di kawasan perioral.
Mereka meliputi kulit kepala, kaki, perineum, dada kanak-kanak. Mereka jarang muncul di tangan dan kaki, berbeza dengan jenis bulosa epidermolisis yang lain. Pengecualian adalah falang terminal jari, di mana kuku berada. Plat itu sendiri runtuh, terkelupas dan hilang selama-lamanya. Semua membran mukus sering terkena ruam.
Erosi sembuh dengan sangat perlahan. Di tempat mereka, atrofi kulit, parut kasar terbentuk. Oleh kerana komplikasi yang timbul, pertumbuhan bayi berhenti. Mereka tidak menambah berat badan. Kanak-kanak seperti itu sering mati sebelum usia tiga tahun kerana jangkitan, keletihan dan gangguan peredaran darah.
Bullosa epidermolisis sempadan sederhana hingga teruk berbeza dari varian sebelumnya dalam kursus yang lebih ringan. Gelembung juga dapat dijumpai pada bayi yang baru lahir, tetapi tidak ada bekas luka ketika sembuh. Ciri khas varian klinikal ini adalah keguguran rambut fokus dan atrofi kulit kepala yang teruk. Bayi seperti itu tumbuh dan berkembang sesuai dengan usia. Penyakit ini tidak mempengaruhi penyakit ini.
Bullosa epidermolisis distrofi
Kumpulan bullosa epidermolisis ini diklasifikasikan kepada dua subkumpulan bergantung kepada jenis pewarisan: autosomal dominan atau autosomal resesif. Ini bermaksud bahawa mutasi genetik diwarisi secara jelas. Dalam kes pertama, untuk perkembangan penyakit ini, hanya satu gen mutan yang cukup, yang ada pada ayah atau ibu anak itu. Dalam kes kedua, kedua ibu bapa mesti menjadi pembawa gen yang rosak, tetapi mereka sendiri akan sihat. Tetapi bayi akan dilahirkan dengan penyakit yang serius.
Bullosa epidermolisis distrofi dominan dicirikan oleh pembentukan lepuh pada semua bahagian badan dan membran mukus sejak lahir. Dalam beberapa kes klinikal, lepuh berkembang walaupun kemudian. Mereka cenderung berulang tetapi cepat sembuh dengan parut dan hipopigmentasi. Bentuk epidermolisis bullosa ini tidak mempengaruhi pertumbuhan dan perkembangan anak.
Bullosa epidermolisis distrofik berulang lebih kerap daripada bentuk penyakit lain menyebabkan kecacatan teruk, walaupun keparahannya mungkin tidak begitu teruk. Lepuh boleh terletak di tangan, kaki, siku, dan lutut, atau merebak ke seluruh badan. Perubahan serupa muncul pada membran mukus. Kerana pembentukan parut kasar, bayi menjadi cacat.
Sindrom Kindler
Kanak-kanak dengan sindrom Kindler dilahirkan dengan lepuh pada kulit dan membran mukus. Pada usia yang lebih tua, fotosensitiviti muncul, pigmentasi pada kulit, bentuk tisu parut, dan kuku terkelupas. Disifatkan oleh kerosakan pada saluran gastrointestinal dan saluran kencing.
Komplikasi bullosa epidermolisis
Perkembangan komplikasi sering berlaku pada kanak-kanak yang menderita bullosa epidermolisis sempadan dan distrofi. Penyakit kumpulan ini berlaku dalam bentuk yang paling agresif.
Ulser dan erosi yang meluas pada kulit dari masa ke masa menyebabkan fakta bahawa ia digantikan oleh tisu parut kasar. Ia dicirikan oleh kepekaan yang lemah, kelembutan yang lemah, kekurangan sebum dan kelenjar peluh. Fungsi kulit hilang, selain itu, parut juga merupakan kecacatan kosmetik yang mencacatkan penampilan.

Penggantian cicatricial pada kulit kelopak mata menyebabkan pembatasan pergerakan mereka. Anak itu tidak akan dapat membuka atau menutup mata sepenuhnya, akibatnya organ penglihatan mungkin dibiarkan tanpa perlindungan. Bentuk mata juga akan berubah sehingga tertutup sepenuhnya. Akibatnya, konjungtivitis, blepharitis dan penyakit radang mata yang lain bergabung. Kanak-kanak itu mungkin hilang sama sekali.
Parut di mulut membawa kepada pembentukan mikrostomi. Celah mulut ditarik bersama, disempitkan. Akibatnya, bayi tidak akan dapat membuka mulutnya hingga akhir. Proses menelan dan pertuturan terganggu.
Tisu parut kasar di kawasan sendi menyebabkan pergerakan terhad di dalamnya. Anak tidak akan dapat membengkok dan meluruskan sendi sepenuhnya, kerana parut tidak akan dapat meregangkan sepenuhnya seperti kulit biasa. Sendi sering berada dalam kedudukan yang sama, kontrakturnya berkembang - kekakuan.
Hakisan dan kelembapan di kawasan jari menyebabkan fakta bahawa tisu parut terbentuk dengan peleburan jari. Dalam kes ini, tangan anak akan hilang fungsi cengkaman.
Erosi pada selaput lendir juga dapat sembuh dengan pembentukan tisu parut, yang menyebabkan penyempitan - penyempitan esofagus, saluran pernafasan dan saluran kencing, usus. Anak tidak dapat menelan makanan sepenuhnya, bercakap, bernafas, buang air kecil, oleh itu, dia akan menurunkan berat badan hingga cachexia, sering jatuh sakit dengan penyakit radang paru-paru dan ginjal. Penyerapan makanan dalam usus akan terganggu. Ini akan memburukkan lagi keadaan bayi.
Ulser dan erosi pada kulit adalah pintu masuk ke pelbagai agen berjangkit. Oleh itu, jika peraturan berpakaian tidak dipatuhi, keradangan kulit bernanah mungkin terjadi, dan kadang-kadang keracunan darah sistemik - sepsis.
Orang dengan epidermolisis bullosa berisiko tinggi terkena barah kulit sel skuamosa.
Jangka hayat dengan epidermolisis bullosa
Jangka hayat bayi rama-rama bergantung kepada bentuk penyakit ini. Prognosis yang paling baik untuk bayi yang menderita bullosa epidermolisis sederhana. Mereka dapat hidup dan belajar sepenuhnya. Terdapat pilihan bahawa penyakit ini biasanya akan hilang seiring bertambahnya usia.
Bullosa epidermolisis sempadan umum dicirikan oleh jalan yang paling teruk. Kanak-kanak dengan bentuk penyakit ini kadang-kadang tidak dapat hidup sehingga tiga tahun kerana perkembangan komplikasi yang tidak sesuai dengan kehidupan.
Dalam bentuk penyakit lain, dalam kebanyakan kes, kecacatan teruk berkembang. Tetapi bayi masih mempunyai peluang untuk menjalani kehidupan yang panjang dengan pengawasan dan penjagaan yang betul.
Diagnostik bulosa epidermolisis
Diagnosis epidermolisis bullosa tidak selalu dapat dibuat berdasarkan aduan. Untuk mengesahkan atau membantahnya, anda memerlukan diagnostik makmal yang kompleks di klinik besar. Analisis genetik juga diperlukan.
Biopsi kulit adalah wajib, selalu dari pundi kencing segar. Sepotong kulit harus segera dibekukan dalam nitrogen cair, atau direndam dalam garam. Untuk penyimpanan jangka panjang, sekeping kulit diletakkan di media pengangkutan khas. Biosampel diperiksa di bawah mikroskop berkuasa khas dengan pemprosesan media kontras. Adalah mungkin untuk menentukan kelainan pada struktur kulit, dan untuk mengenal pasti kekurangan komponen protein tertentu.
Sebelum bayi dilahirkan, analisis genetik memainkan peranan penting. Ia tidak mempengaruhi rawatan, tetapi diperlukan oleh keluarga di mana anak dengan epidermolisis bullosa sudah dibesarkan, dan di mana kehamilan kedua dirancang.
Prinsip rawatan untuk epidermolisis bullosa
Di dunia moden, epidermolisis bullosa dianggap sebagai penyakit yang tidak dapat disembuhkan. Tindakan rawatan dan pencegahan harus mencegah trauma pada kulit.Untuk ini, kulit "anak rama-rama" memerlukan penjagaan yang betul. Untuk keselesaan pesakit muda, penting untuk menghilangkan rasa gatal dan sakit.
Untuk mengelakkan komplikasi yang mengancam nyawa, perlu menangani jangkitan kulit tepat pada masanya. Sekiranya komplikasi timbul dari sistem pencernaan dan sendi, ia perlu diperbaiki tepat pada waktunya. Ibu bapa bayi rama-rama harus dilatih untuk merawatnya secepat mungkin. Baginya, plester lembut khas, pembalut, salap dengan perak, antiseptik digunakan.

Gelembung besar biasanya pecah dengan lembut. Ejen yang diperlukan digunakan pada hakisan yang dihasilkan. Kemudian luka ditutup dengan pembalut. Mereka menggunakan pembalut atraumatik khusus yang mengandungi agen sorben, antiseptik, regenerasi dan antimikroba.
Filem hidrogel dan hidrokoloid khas juga telah dikembangkan. Mereka menutup hakisan dan ulser, sehingga mencegah pengeringan. Sponge kolagen boleh digunakan. Mereka boleh melekat erat pada luka, dan berpisah dengan sendirinya jika basah atau sembuh sepenuhnya.
Pembalut kedua digunakan di atas. Terima kasih kepadanya, pembalut perubatan akan terpasang dengan baik dan akan dipasang dengan kuat di tempatnya. Pembalut fiksasi tidak boleh menekan kulit dengan kuat dan diikat pada simpul yang ketat agar tidak mencederakan kulit. Untuk mengelakkan komplikasi, pembalut yang terpisah untuk setiap jari kanak-kanak diperlukan. Anggota badan semasa membalut tidak boleh dibengkokkan atau tidak bengkok. Berpakaian bayi dengan varian klinikal yang teruk dari epidermolysis bullosa boleh memakan masa 1 - 2 jam.
Bayi yang menderita bullosa epidermolisis harus diperhatikan oleh banyak pakar dan diperiksa secara berkala oleh pakar dermatologi, pakar pediatrik, ahli gastroenterologi, pakar otolaryngologi, pakar bedah. Sekiranya perlu, rundingan pakar bedah toraks dan plastik diperlukan.
Dalam beberapa kes, perlu diberikan antibiotik dalam suntikan. Anak harus menerima semua nutrien dan vitamin yang diperlukan dengan makanan. Kanak-kanak diberikan campuran protein tinggi khusus. Sekiranya, kerana komplikasi, bayi tidak dapat menelan makanan secara normal, gastrostomi mungkin dipasang.
Untuk membantu ibu dan bapa bayi istimewa, dana "anak rama-rama" telah dibuat. Dia membantu dalam rawatan dan pemulihan kanak-kanak yang menderita epidermolisis bullosa, membeli ubat untuk mereka. Sekiranya bantuan psikologi dan undang-undang diperlukan, ia juga akan diberikan terima kasih kepada dana. Pakarnya juga membantu doktor yang menghadapi penyakit.
Kesimpulannya
Epidermolysis bullosa adalah penyakit yang serius dan, malangnya, tidak dapat disembuhkan. Semua aktiviti rawatan hanya bersifat paliatif. Tetapi dengan terapi yang dimulakan dengan betul dan tepat pada masanya, anda dapat mengurangkan penderitaan bayi dengan ketara dan memberi dia peluang untuk hidup panjang.